科学研究



对瞬态反应中间体(如·OH等活泼自由基)实现时空控制,是多相催化领域在复杂介质中精准转化的核心挑战。非选择性扩散从根本上限制了催化体系在复杂介质中的效率。高活性自由基一旦生成,便不可避免地发生非选择性迁移,导致大量非特异性副反应。如何解耦催化剂的本征活性与目标选择性,将短寿命中间体严格限域在预设空间内供特定底物即时消耗,是该领域亟待突破的关键科学问题。
近日,william威廉中文赵国华教授课题组受自然界酶催化中“邻近效应”的启发,设计了一种适配体功能化的Au-Fe双单原子催化剂(Apt-Au1-Fe1/NC),通过将原子级分散的金属位点与识别元件相集成,构筑了一个“智能反应口袋”,实现了对目标底物的时空可控选择性转化。相关成果以“Dual-Single-Atom Catalyst with Aptamer-Engineered Confined Reactive Milieu for Spatiotemporally Controlled Selective Conversion”为题,发表于国际材料领域顶级期刊《先进材料》(Advanced Materials, 2026, e73645)。

针对活泼自由基难以精准利用的难题,研究团队在原子尺度上设计了一个限域反应微环境。其中,Fe单原子通过三电子氧还原原位生成·OH,而相邻Au原子作为“分子锚点”固定适配体,预富集目标底物。双原子紧密排列使生成区与识别区相邻甚至重叠,构筑了富含·OH的纳米“反应口袋”。该结构模拟了天然多酶复合物中的“底物通道”,确保·OH在扩散前即被捕获的底物即时消耗,为实现精准时空可控的选择性转化奠定了基础。
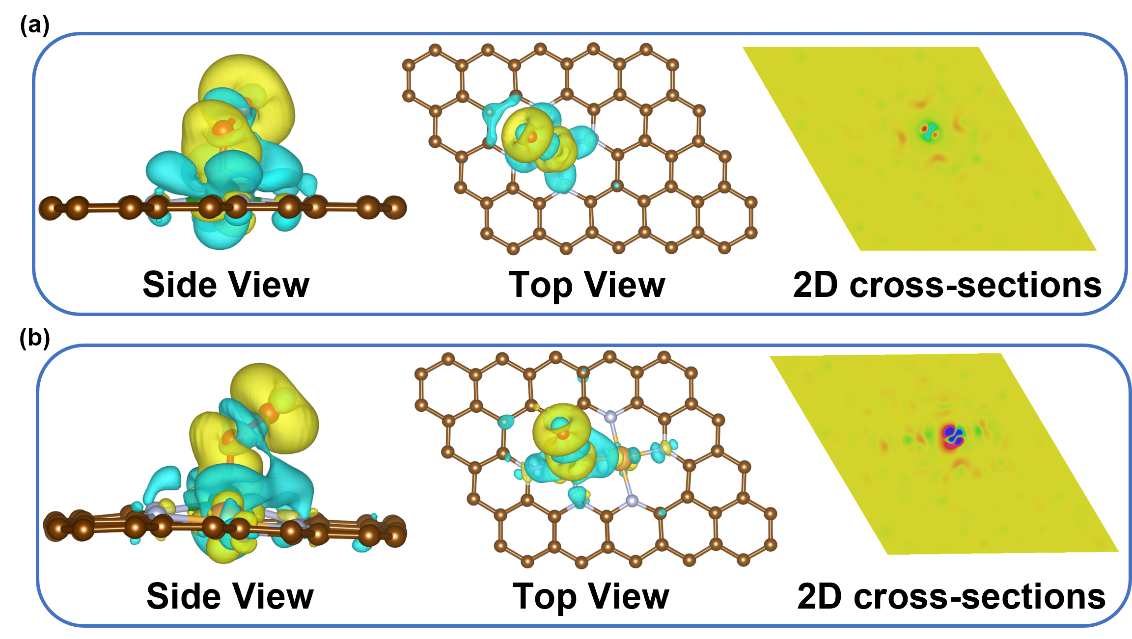
电化学测试表明,引入Au单原子后催化剂起始电位正移、极限电流密度提升,证实Au对Fe-N4位点的电子结构调控增强了本征ORR活性。接枝适配体后性能未明显改变,实现了“识别”与“催化”的协同兼容。旋转环盘电极测试显示,Apt-Au1-Fe1/NC的电子转移数稳定在3.0附近(对照样品约为3.5),证实Au-Fe双位点实现了高效3e- ORR过程,原位生成·OH。理论计算揭示了Au-Fe双位点的协同效应。Fe-N4-C的电荷重分布局域在Fe位点,而Au1-Fe1/NC中出现从Au-Fe双原子向O2的显著电荷转移,将O-O键从1.23 Å拉长至1.42 Å,有效削弱了O-O键。该协同机制中,Fe位点吸附O2,相邻Au原子调控Fe 3d轨道并稳定*OOH中间体,将决速步能垒降至0.424 eV,并将反应锁定在3e- ORR路径上,为构筑限域·OH反应微环境奠定了基础。
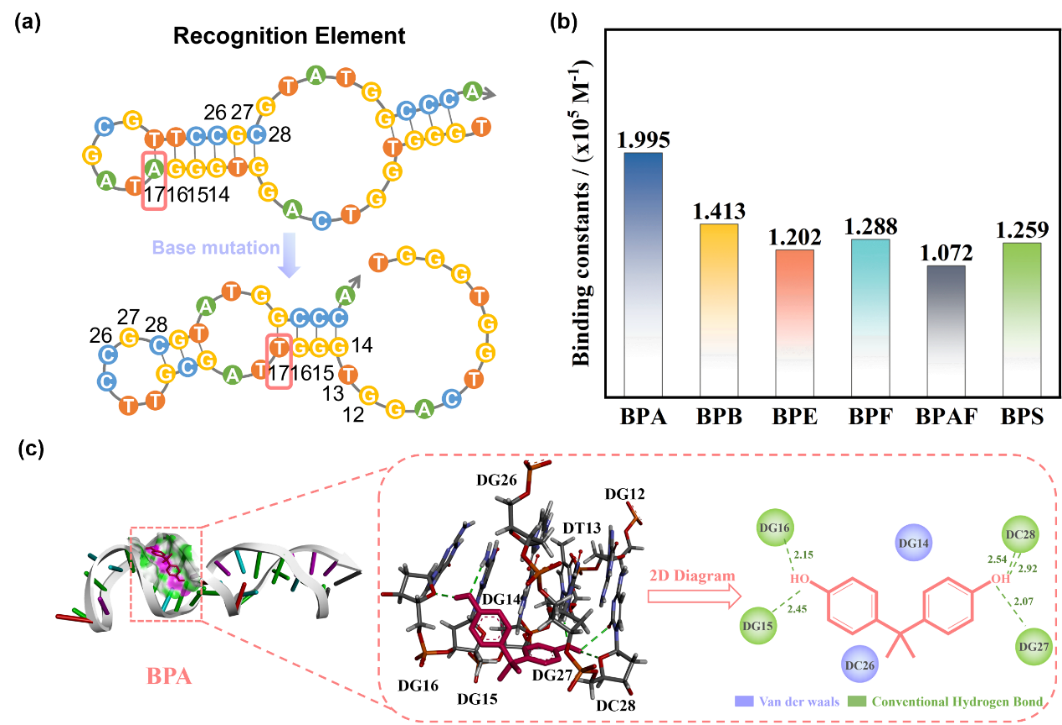
在确认催化剂具备高效·OH生成能力之后,研究团队进一步赋予其精准的分子识别功能。原始适配体仅识别双酚A(BPA),通过定点诱变将第17位腺嘌呤(A)突变为胸腺嘧啶(T),构象重排后形成了通用结合口袋。结合常数计算表明,突变后适配体对BPA保持高亲和力(Ka = 1.995×105 M-1),对另外五种双酚类的亲和力也达到1.07-1.41×105 M-1,实现了广谱识别。分子对接模拟揭示,范德华力、氢键及π-π堆积的多重协同作用,使适配体作为“限域识别单元”在微环境内高效捕获底物,实现了靶向识别。
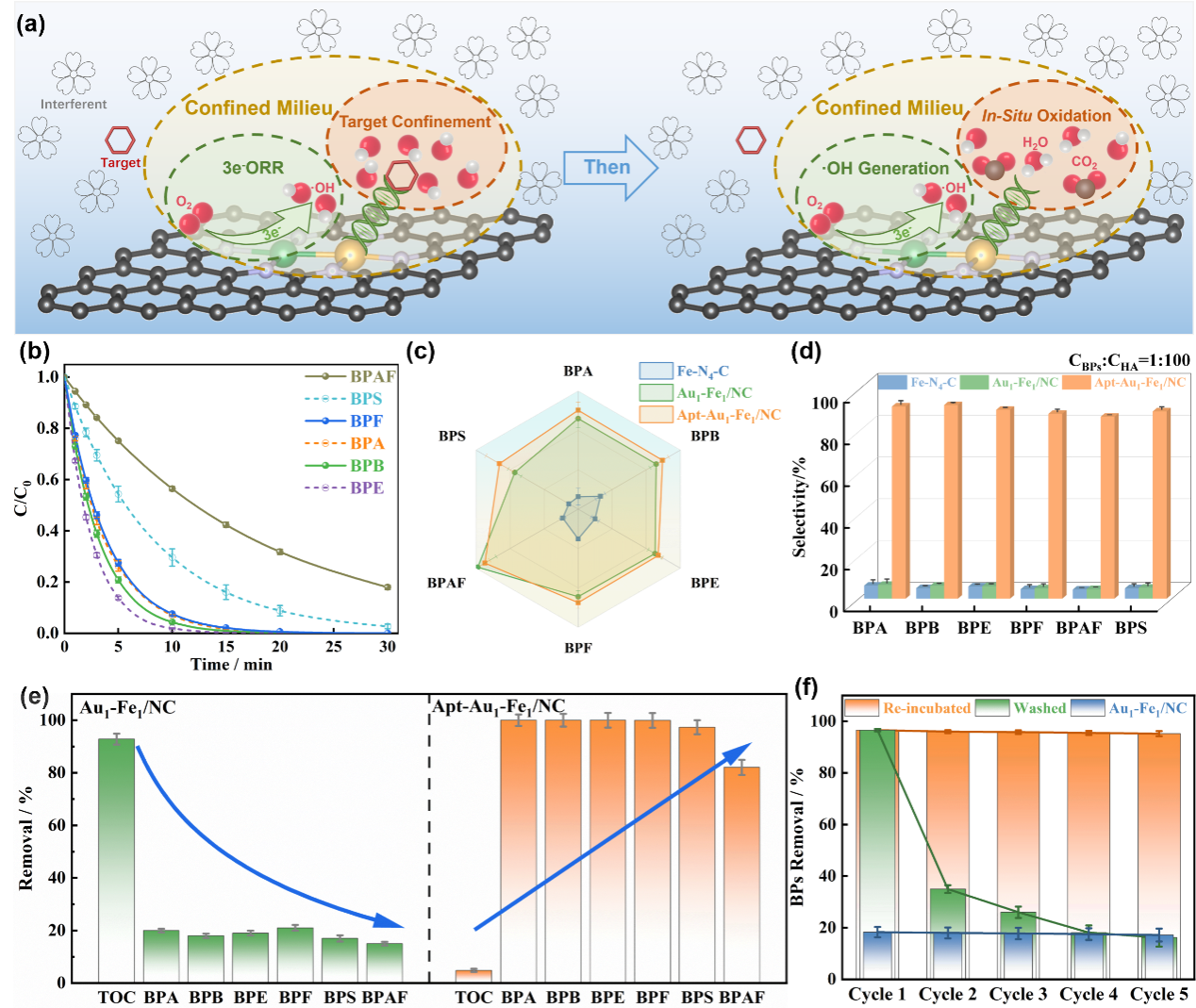
在构筑限域反应微环境的基础上,研究团队验证了该体系对目标底物的时空可控选择性转化能力。适配体作为“识别单元”将目标底物严格限域在·OH附近,防止其非选择性耗散。在20分钟内对目标底物降解率超过95%,在混合底物中选择性系数高达87%,而无适配体体系均低于10%。背景基质中TOC去除率仅5%(对照高于90%)。通过适配体重新孵育的动态再生策略,体系在五个循环中去除率始终高于95%。更换适配体后,该策略对土霉素(88.26%)和邻苯二甲酸二丁酯(87.54%)同样有效。
该工作通过在无机活性中心上施加生物识别单元的空间约束,成功解耦了活性与选择性,建立了“限域反应微环境工程”新范式。通过更换适配体,该策略可推广至有机污染物降解、精准合成、不对称催化及选择性官能团转化等领域,为复杂化学环境中时空可控选择性转化提供了通用设计策略。
上述研究工作得到了国家重点研发计划、国家自然科学基金和WilliamHill中文医学-X交叉学科研究等项目支持。赵国华教授为论文的通讯作者,博士生刘景妍为论文第一作者。
原文链接:https://doi.org/10.1002/adma.73645

Copyright © 威廉希尔williamhill(中国区)中文官方网站-主頁欢迎您 版权所有